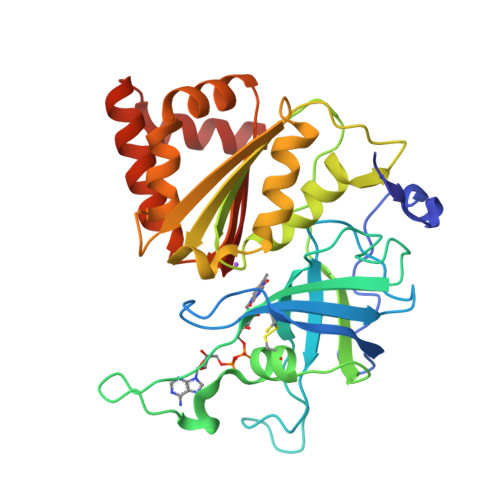
Using a conformation-dependent stereochemical library improves crystallographic refinement of proteins.
Tronrud, D.E., Berkholz, D.S., Karplus, P.A.(2010) Acta Crystallogr D Biol Crystallogr 66: 834-842
- PubMed: 20606264
- DOI: https://doi.org/10.1107/S0907444910019207
- Primary Citation of Related Structures:
3LO8 - PubMed Abstract:
The major macromolecular crystallographic refinement packages restrain models to ideal geometry targets defined as single values that are independent of molecular conformation. However, ultrahigh-resolution X-ray models of proteins are not consistent with this concept of ideality and have been used to develop a library of ideal main-chain bond lengths and angles that are parameterized by the phi/psi angle of the residue [Berkholz et al. (2009), Structure, 17, 1316-1325]. Here, it is first shown that the new conformation-dependent library does not suffer from poor agreement with ultrahigh-resolution structures, whereas current libraries have this problem. Using the TNT refinement package, it is then shown that protein structure refinement using this conformation-dependent library results in models that have much better agreement with library values of bond angles with little change in the R values. These tests support the value of revising refinement software to account for this new paradigm.
Organizational Affiliation:
Department of Biophysics and Biochemistry, Oregon State University, Corvallis, Oregon 97331, USA.