Crystal Structure of Human T-protein of Glycine Cleavage System at 2.0A Resolution and its Implication for Understanding Non-ketotic Hyperglycinemia
Okamura-Ikeda, K., Hosaka, H., Yoshimura, M., Yamashita, E., Toma, S., Nakagawa, A., Fujiwara, K., Motokawa, Y., Taniguchi, H.(2005) J Mol Biology 351: 1146-1159
- PubMed: 16051266
- DOI: https://doi.org/10.1016/j.jmb.2005.06.056
- Primary Citation of Related Structures:
1WSR, 1WSV - PubMed Abstract:
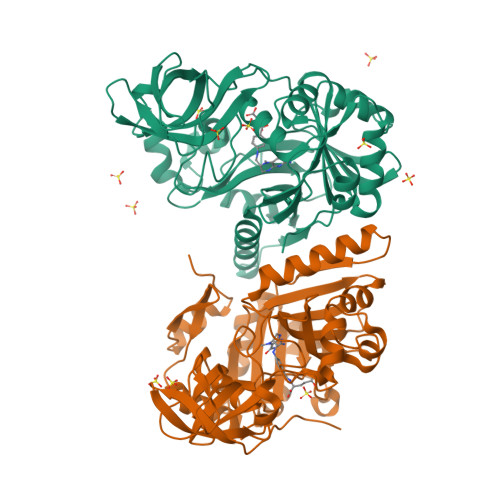
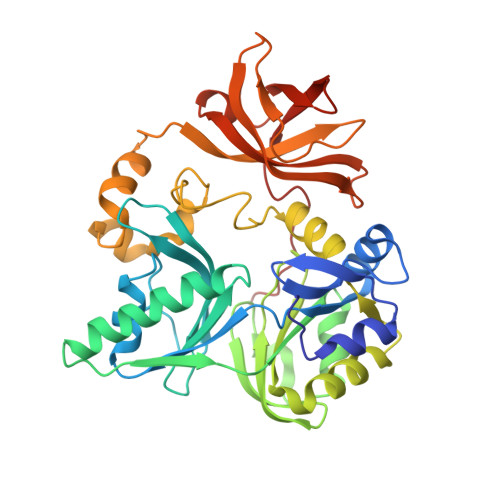
T-protein, a component of the glycine cleavage system, catalyzes the formation of ammonia and 5,10-methylenetetrahydrofolate from the aminomethyl moiety of glycine attached to the lipoate cofactor of H-protein. Several mutations in the human T-protein gene cause non-ketotic hyperglycinemia. To gain insights into the effect of disease-causing mutations and the catalytic mechanism at the molecular level, crystal structures of human T-protein in free form and that bound to 5-methyltetrahydrofolate (5-CH3-H4folate) have been determined at 2.0 A and 2.6 A resolution, respectively. The overall structure consists of three domains arranged in a cloverleaf-like structure with the central cavity, where 5-CH3-H4folate is bound in a kinked shape with the pteridine group deeply buried into the hydrophobic pocket and the glutamyl group pointed to the C-terminal side surface. Most of the disease-related residues cluster around the cavity, forming extensive hydrogen bonding networks. These hydrogen bonding networks are employed in holding not only the folate-binding space but also the positions and the orientations of alpha-helix G and the following loop in the middle region, which seems to play a pivotal role in the T-protein catalysis. Structural and mutational analyses demonstrated that Arg292 interacts through water molecules with the folate polyglutamate tail, and that the invariant Asp101, located close to the N10 group of 5-CH3-H4folate, might play a key role in the initiation of the catalysis by increasing the nucleophilic character of the N10 atom of the folate substrate for the nucleophilic attack on the aminomethyl lipoate intermediate. A clever mechanism of recruiting the aminomethyl lipoate arm to the reaction site seems to function as a way of avoiding the release of toxic formaldehyde.
Organizational Affiliation:
Institute for Enzyme Research, The University of Tokushima, Tokushima 770-8503, Japan. ikeda@ier.tokushima-u.ac.jp