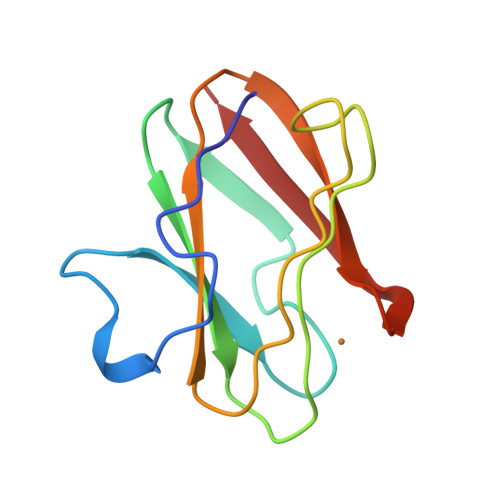
X-ray structure of the cupredoxin amicyanin, from Paracoccus denitrificans, refined at 1.31 A resolution.
Cunane, L.M., Chen, Z.W., Durley, R.C., Mathews, F.S.(1996) Acta Crystallogr D Biol Crystallogr 52: 676-686
- PubMed: 15299631
- DOI: https://doi.org/10.1107/S0907444996001072
- Primary Citation of Related Structures:
1AAC - PubMed Abstract:
High-resolution X-ray diffraction data to d(min) = 1.31 A were collected on a Xuong-Hamlin area detector from crystals of the blue-copper protein amicyanin, isolated from P. denitrificans. With coordinates from the earlier 2.0 A structure determination as a starting point, simulated annealing and restrained positional and temperature factor refinements using the program X-PLOR resulted in a final R factor of 15.5%, based on 21 131 unique reflections in the range 8.0-1.3 A. Comparison of the 1.31 A structure with that at 2.0 A shows the same basic features. However, the high-resolution electron-density maps clearly reveal additional solvent molecules and significant discrete disorder in protein side chains and within the solvent structure. As a consequence of modelling side-chain disorder, several new hydrogen-bonding interactions were identified.
Organizational Affiliation:
Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, MO 63110, USA.